Single-cell submission guide
Start your single-cell submission using the single cell sequencing template in Annotare. Follow the guides below for information about data formats and what metadata to include. Note that larger experiments with more than 1000 raw data files in one experiment are started in Annotare but will be completed manually with the help of a curator.
Sample information
What is a "sample" in a single-cell experiment?
The general rule is to create and annotate 1 sample per library. In plate-based methods, like Smart-seq2, one library will correspond to a single cell, which will be the sample. In case you have multiplexed data from droplet-based platforms like 10x, one library will contain several thousands of individual cells. The following attributes are suggested for single cell experiments and can be removed if they don't apply.
Single cell level attributes
-
Inferred cell type
If your experiment involves discovering new cell types or you work with pooled cells from a whole organ or organism, the precise cell type might be undefined at the start of the experiment but inferred later during the analysis. In that case, annotate the samples with the inferred cells type using the attribute "inferred cell type". The attribute "cell type" should refer to the input cell type that is known at the start of the experiment.
Note: If you have multiplexed data and cannot enter this in the samples table, please include the annotations in a separate processed data file (see below).
Example: E-MTAB-5953 -
Single cell identifier (experimental variable)
If the purpose of your experiment is comparing the heterogeneity within one cell population, you might not have any other experimental variables. In this case, you can use the term "single cell identifier" as experimental variable and enter the cell IDs as values. Example: E-MTAB-6142 -
Single cell well quality (pre-analysis)
If your single-cell experiment included visual inspection of the cells before lysis, you can include this information in the sample attributes as "single cell well quality". The values for this attribute should be "OK" or "not OK" (i.e. the well should be discarded from data analysis). If you have more detailed information about the "bad" wells, the following terms should be used:- "debris"
- "multiple cells" or number of cells if known
- "dead cell"
- "no cell"
-
Post-analysis well quality
You might have quality information about the dataset, e.g. which cells were excluded from your analysis due to insufficient sequencing quality. You can use the attribute "post analysis single cell quality" and annotate each cell with "pass" or "fail". Example: E-MTAB-5522
Examples of single-cell sample attributes
| Example | single cell identifier (experimental variable) | inferred cell type | single cell well quality | post-analysis single cell quality |
|---|---|---|---|---|
| Sample 1 | cell 1 | cell type A | OK | pass |
| Sample 2 | cell 2 | cell type B | OK | pass |
| Sample 3 | cell 3 | not applicable | OK | fail |
| Sample 4 | cell 4 | cell type C | OK | pass |
| Sample 5 | cell 5 | not applicable | 2 cells | fail |
| Sample 6 | cell 6 | not applicable | debris | fail |
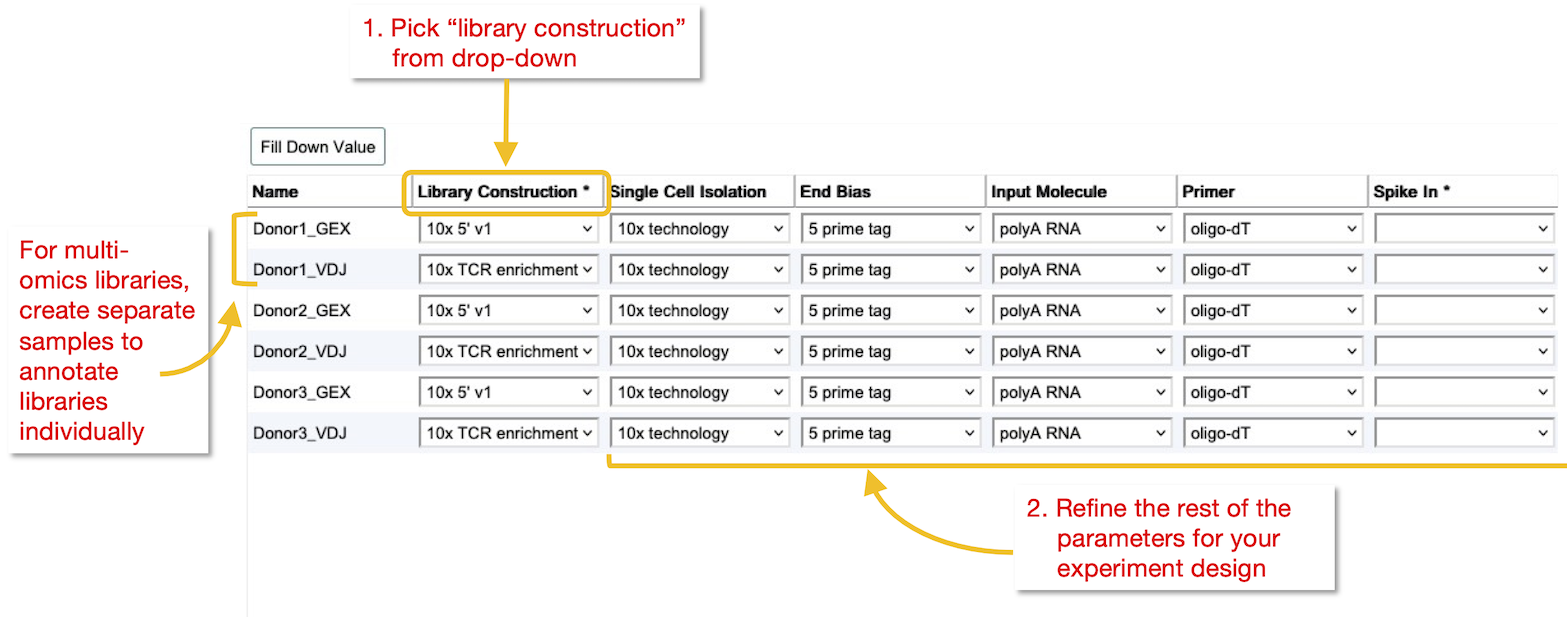
Single cell library information
1. Pick library construction method
Select the type of single cell library construction that you followed from controlled vocabulary drop-down menu. For some of the most commonly used methods, this will pre-fill the other fields with the default values. These can still be adjusted afterwards.
2. Refine parameters
Check each parameter carefully, selecting the suitable option from the drop-down menu.
General description
-
Single Cell Isolation
The method used for selection of single cells, to allow individual cell barcoding -
End Bias
The end of the nucleic acid molecule that is preferentially sequenced (bias in read distribution) -
Input molecule
The type or fraction of nucleic acid that was captured in the library -
Primer
The type of primer used for reverse-transcription
Spike-in RNAs
-
Commercial spike-ins
If you have used spike-in that are commercially available, please include the kit name, catalogue number and dilution in the library construction protocol. -
Custom spike-ins
If you have used a non-commercial spike-in set, describe this in the library construction protocol. Additionally, upload a table (tab-delimited text) with the names and concentrations of the spike-ins, and a fasta file containing the nucleotide sequences. Example: E-MTAB-3624.additional.1.zip -
Spike-in annotation
You can include spike-in information at the cell level in the sample annotation under "spike in" and "spike in dilution".Example of spike-in annotation
Example spike in spike in dilution Sample 1 ERCC mix1 1:40000 Sample 2 ERCC mix2 1:40000 Sample 3 ERCC mix1 and mix2 1:40000
Unique Molecular Identifiers
-
If your single-cell protocol uses unique molecular identifiers (UMIs), all relevant information about the UMI barcodes should be included to enable re-analysis of the data. We suggest to include the following:
- UMI barcode read (the file that contains the UMI). Values: read1/read2/index1/index2
- UMI barcode offset (start position of UMI barcode in the sequence). Values: (number, 0 for start of read)
- UMI barcode size (length of UMI barcode in bp). Values: (number)
3. Define library layout for multiplexed data
For 10x technology, ensure you have selected the correct version of the 10x chemistry, e.g. v2 or v3. Note that Annotare currently only provides templates for gene expression experiments.
For other large-scale single-cell sequencing methods or where modifications were done to the standard 10x protocol, please include specifications about the multiplexing and barcodes. We suggest to include the following to specify the positions and size of the barcodes:
| Attribute | Description | Possible values |
|---|---|---|
| cDNA read | the file that contains the cDNA read | index1/index2/read1/read2 |
| cDNA read offset | offset in sequence for cDNA read (in bp) | (number, 0 for start of read) |
| cDNA read size | length of cDNA read (in bp) | (number) |
| UMI barcode read | the file that contains the UMI barcode read | index1/index2/read1/read2 |
| UMI barcode offset | offset in sequence for UMI barcode read (in bp) | (number, 0 for start of read) |
| UMI barcode size | length of UMI barcode read (in bp) | (number) |
| cell barcode read | the file that contains the cell barcode read | index1/index2/read1/read2 |
| cell barcode offset | offset in sequence for cell barcode read (in bp) | (number, 0 for start of read) |
| cell barcode size | length of cell barcode read (in bp) | (number) |
| sample barcode read | the file that contains the sample barcode read | index1/index2/read1/read2 |
| sample barcode offset | offset in sequence for sample barcode read (in bp) | (number, 0 for start of read) |
| sample barcode size | length of sample barcode read (in bp) | (number) |
File types and formats
Raw data
In addition to the rules below, all raw data must conform to the general accepted sequencing file formats.
Raw read data should be submitted in fastq.gz format. Prepare one file per cell (or 2 if you have used paired-end sequencing), following the general recommendations from the European Nucleotide Archive (ENA). An exception are droplet-based technologies like 10x and Drop-seq and other methods for which data is not demultiplexed, where we allow submission of multiplexed data. A few extra rules apply here.
For 10x technology, please provide fastq.gz files, for example as generated by CellRanger software from bcl files. These are usually 2-4 fastq.gz files per library, containing the cDNA read and several barcode reads in known positions.
We can currently validate files that follow the 10x file naming conventions:
- R1 + R2 (+ I1)
- R1 + R2 + R3 + I1
- RA + I1 + I2
For other formats where the data cannot be represented as 2 paired-end fastq files, please convert your raw data to unaligned BAM format. We recommend using the following SAM specification tags for the typical single cell barcode sequences:
- BC: Sample barcode sequence
- QT: Sample barcode quality
- CR: Cell barcode sequence
- CY: Cell barcode quality
- RX: UMI barcode sequence
- QX: UMI barcode quality
Processed data
Processed data is welcome in addition to raw data. Most commonly this would be a raw or normalised read count matrix. The format of any matrix file should be tab-delimited text. We also accept other commonly generated formats such as sparse matrix files. Also other types of analysis result files can be included, e.g. alignment files, cluster or cell type annotations.
Inferred cell type annotations (see above) are a very important piece of metadata that greatly enhance the value of single-cell sequencing data stored in ArrayExpress and we very much encourage all submitters to add this information to their submission. Since information about individual cells can't be entered at the sample annotation or file level for multiplexed data, please include inferred cell type annotations and any other cell-level annotations as a processed data file. Any such file should at least contain the following information, so the correct connection between the annotations and the the relevant samples and sequencing reads can be made:
| Column header | Library ID/Sample ID | Cell Barcode | Inferred cell type |
| Description: | This should match with the raw data file prefix | The sequence of the cell barcode | assigned cell type annotation |
| Example: | Sample1_run1 | ATGGTCATCGT | macrophage |
Additional files
It is encouraged to upload any additional files that facilitate data analysis, e.g. text files containing lists of known barcodes in the library. These files will be linked to the experiment and don't need to be associated with any specific sample in the submission. Please leave a description of such files in an appropriate protocol, e.g. the normalisation data transformation protocol. Example: E-MTAB-6153_sample_barcodes.txt
Cell hashing and cell surface antibody readout are other areas where additional files are useful, as there is no standard format for encoding this in Annotare (yet). For cell hashing an additional table is needed with the individual sample annotations and respective barcodes. For cell surface antibody readout, such as CITE-seq, the file should include the antibody targets and their barcode sequence. Example: E-MTAB-9295_features.txt
Split-seq/Parse Evercode: In general, one sample should correspond to one sequencing library, and each raw data file should belong to a single sample/library. For Parse Evercode or Split-seq data, create a separate sample in Annotare for each sublibrary and assign only the matching FASTQ file pair to that sample. If a sublibrary was sequenced across multiple lanes, additional FASTQ pairs can be added to the same sample. FASTQ files cannot be assigned to all samples. Please provide a separate mapping spreadsheet containing biological sample metadata and corresponding barcodes, and assign these as processed data files. The same mapping file can be linked to multiple samples.
Note: Currently there is no dedicated way to assign "additional files" in Annotare. Please use the "Processed Data" columns in the file assignment to attach such files to your submission.
Protocols
Sample collection protocol
-
Method of cell singularisation
Please include a description of how the cells were treated and separated into single-cells, e.g. FACS or microfluidics (Fluidigm).
Library construction protocol
-
Single-cell library construction
Please mention the type of single-cell library method that was used (e.g. Smart-seq2, 10x, Drop-seq) and give any relevant literature references. -
Library construction kit
Please include the name, manufacturer and catalogue number of the library preparation kit(s) that were used.
Sequencing protocol
-
Technical replicates
Please include details about technical replicates, e.g. if the same libraries were sequenced multiple times or across several lanes.