Sequencing library information
The following information is required by the European Nucleotide Archive (ENA) that stores that raw data files of your submission. The information helps to correctly interpret the submitted data. Controlled vocabulary is provided in drop-down lists where possible.
For all sequencing experiments:
- Library Layout: whether to expect SINGLE or PAIRED end reads.
- Library Source: the type of source material that is being sequenced.
- Library Strategy: the sequencing technique intended for the library.
- Library Selection: the method used to select and/or enrich the material being sequenced.
For paired-end sequencing experiments only:
- Nominal Length: the expected size of the insert. The insert being
the fragment sequenced, as chosen from size fractionation (e.g. cutting out a band from an agarose gel). If nominal
length is 500bp, put down "500" as the value. No decimals or ranges (e.g. 100-200) allowed, and it cannot be zero. This information guides sequence aligner algorithms to place the two mates of a pair of reads on the genome sequences separated by a reasonable distance. Therefore, the nominal length value does not need to be absolutely precise, but also should not be an order of magnitude off:
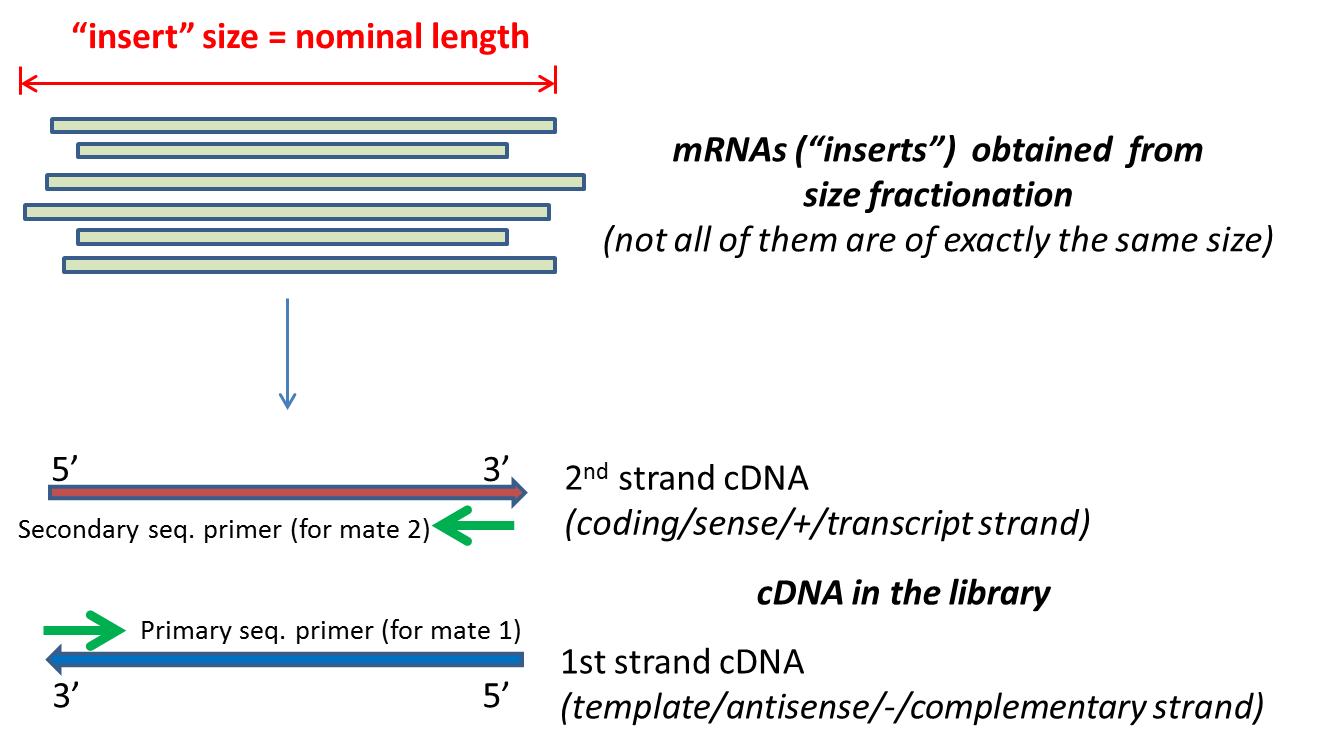
- Nominal SDev: the standard deviation of the nominal length. Decimals are allowed (e.g. 56.4) but no ranges (e.g. 34.5-42.6).
- Orientation: the orientation of the two reads. "5'-3'-3'-5'" for forward-reverse pairs, "5'-3'-5'-3'" for forward-forward pairs.
For strand-specific sequencing experiments:
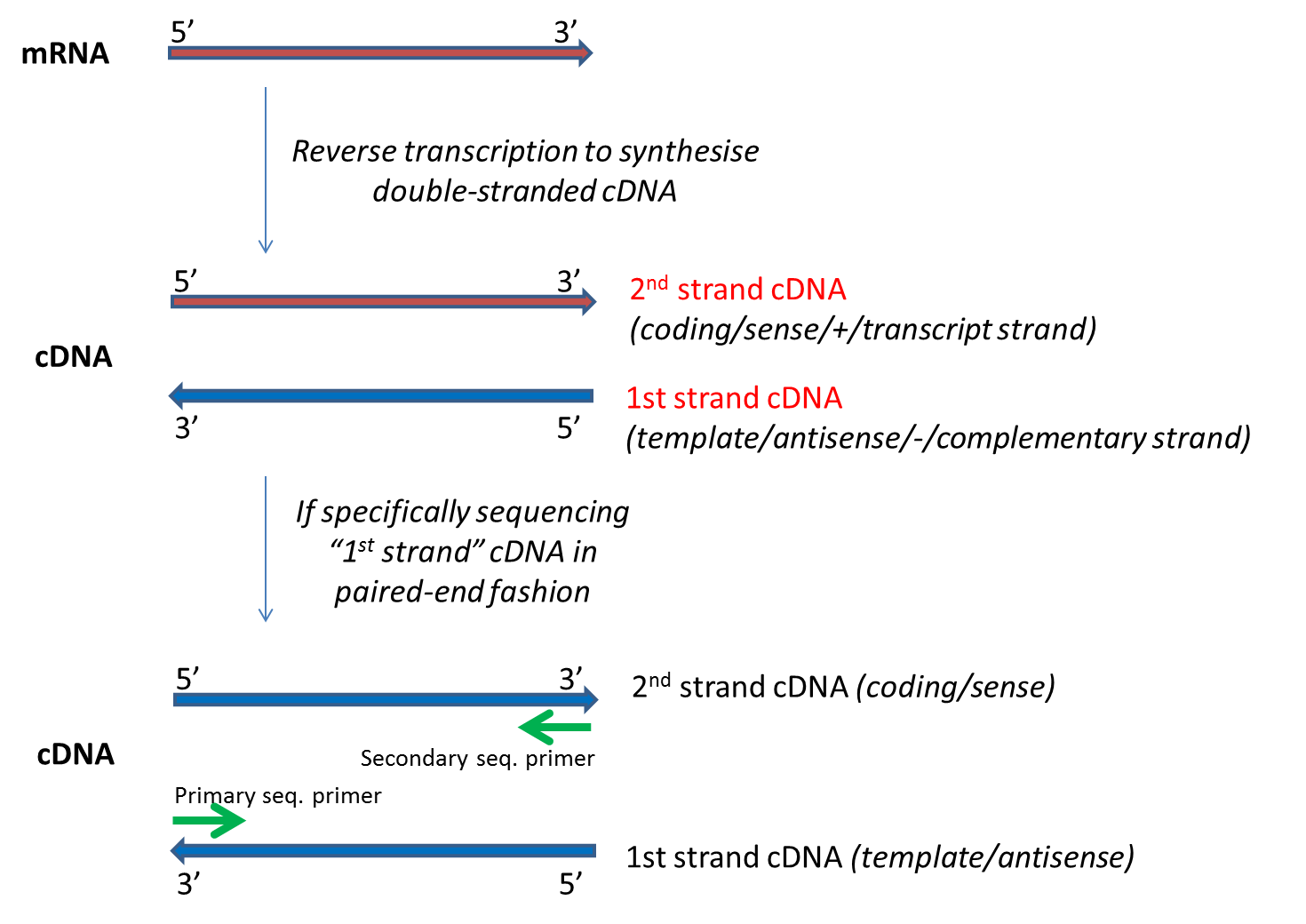
- Library Strand: whether the 'first strand' or 'second strand' of cDNA was used to prepare the library. Note this should not be confused with the forward and reverse strands one would get in a paired-end sequencing reaction.
"First strand" corresponds to the "template"/ "antisense" / "-" strand and has the reverse complement sequence of the transcribed mRNA, wherease "second strand" corresponds to the "coding" /"sense" /"+" strand which has the same sequence as the RNA that is transcribed. The commonly used dUTP-based protocol sequences the 'first strand'. For paired-end strand-specific sequencing studies, sequencing the 'first strand" is sometimes referred to as "mate2_sense" (i.e. mate 2 of the paired-end reads comes from the sense strand). Please see the diagram below for further information: